


-
西伯利亚白刺(Nitraria sibirica Pall)系蒺藜科白刺属植物,为第三纪孑遗植物,分布于蒙古、中亚以及我国西北、华北、东北的沙地、盐碱地地区[1]。西伯利亚白刺具耐盐碱、抗风沙等特性,能在沙漠盐碱等恶劣环境下生存,是一种优良的沙地、盐碱地改良物种,其果实富含多种氨基酸、糖类、黄酮等物质[2-4],营养价值丰富,其地上部分也可作为牲畜饲料。因此,西伯利亚白刺兼有生态和经济价值,有较好的开发利用前景。
目前,关于西伯利亚白刺的研究主要集中在繁殖技术优化[5-6]、果实成分测定[7-8]及生理生化测定[9-11]等方面,有关西伯利亚白刺的分子生物学方面的研究较少[12],基因组学方面的研究也尚未见报道。宏观的研究只能从表层揭示西伯利亚白刺抗逆适应现象 ,并不能从内部机制、进化等层面解释西伯利亚白刺抗逆机理,而全基因组测序可以获取典型基因组特征并获得大量基因序列,对于剖析其生长、发育、抗逆等机理,发掘西伯利亚白刺的生态和经济价值有积极意义[13-14]。
全基因组调查通过了解待测生物基因组的基本特征,可以对全基因组测序组装难度、组装时间和成本等作出大致的评估并作出相应的测序策略调整,是基因组测序前必不可少的步骤之一。
流式细胞术是一种快速预测基因组大小的技术,它通过比较待测植物和标定植物细胞悬液荧光吸收峰相对比值,再根据标定植物的基因组大小来计算待测植物基因组大小[15]。而随着基因组测序技术的成熟及成本的下降,通过全基因组survey来探究待测植物的基因组基本特征不失为一种有效的方法,作为近年来发展较快的基因组预测技术,全基因组survey可以对生物的基因组基本特征测定评估[16-17],相比于流式细胞术等基因组大小预测方法,不仅可以精准预测基因组大小,还可以对基因组复杂程度、杂合率、重复序列比例等有相应的评估,更能切合生物的基因组特征,因而有更好的参考价值。
SSR分子标记以其高重复性、高多态性、共显性遗传、丰度高等优良特性成为了研究群体遗传学、遗传变异和标记辅助选择的有力工具,对于了解西伯利亚白刺的进化有积极的作用。
本研究基于流式细胞术和全基因组survey测序的方法对西伯利亚白刺基因组大小、复杂程度、杂合率等基因组特征有一个较为详细的评估,同时也对其测序方案的制定提出建议,为后续西伯利亚白刺基因组组学研究奠定了良好的基础。
-
将取自内蒙古磴口的野生西伯利亚白刺种子置于4℃下沙藏30 d,置于萌发盒上进行萌发,再将发芽的种子定植于7 cm×7 cm的塑料花盆中(基质配方为河沙∶营养土=1∶1,并在其中掺入少量珍珠岩和蛭石),幼苗生长2个月后取嫩叶备用。流式标定植物为Jaroslav Dolezˇel博士惠赠的番茄‘Stupicke´ polnı´ rane´’ 32品种。
-
使用BD公司influx型号流式细胞仪对西伯利亚白刺基因组大小进行分析,选用mG解离液对植物叶片进行解离,使用碘化丙啶(PI)溶液为荧光染料,采用本番茄作为内标,使用Influx自带分析软件FACSTM分析基因组大小。
操作步骤:于塑料皿上滴加1.5 mL mG解离液,分别取0.5 g西伯利亚白刺、番茄新鲜叶片用刀片迅速切碎后过400目滤网,将收集的滤液1 500 rpm,离心6 min,吸除上清液后重新加入500 μL预冷的mG解离液,加入PI染色液,最后加入10 μg·mL−1的Rnase,避光4℃孵育5 min后低速上机检测。
C值计算公式:C待测样本=C标定×(G0/G1待测样本/G0/G1标定)
式中:G0/G1为流式荧光吸收强度。
mG解离液配方:
45 mmol·L−1 MgCl2,20 mmol·L−1 MOPS,30 mmol·L−1 Na3C6H5O7·2H2O,1%(w/v)PVP-40,0.2%(v/v)TritonX-100,10 mmol·L−1 Na2EDTA,20 μL·mL−1β-巯基乙醇,调节pH至7.0,−20℃下保存。PI为碘化丙啶,使用时至终浓度为50 μg·μL−1,4℃保存。
-
采用CTAB法对西伯利亚白刺的新鲜叶片进行DNA提取,得到的DNA样品用紫外分光光度计检测其浓度、OD260/OD280,再经1%琼脂糖凝胶电泳检测其完整性(电泳条件为:电压180 V,电泳时间:30 min)。
-
检测合格的DNA样品通过Covaris超声波破碎仪打断成片段,并进行末端修复,加poly-A尾,加测序接头,纯化,PCR扩增等步骤后,构建出350 bp双端PE150待测序文库。文库通过Illumina Hiseq平台进行双端PE测序。
-
采用K-mer分析策略,若每条序列的长度为L,K-mer长度为K,可以得到L-K+1个K-mer,再通过这些数据来对基因组大小进行预估,通过Lander-waterman算法对西伯利亚白刺基因组大小进行估计,满足公式:
$ \begin{split} & {{{\rm{C}}_{{\rm{base}}}}{\rm{ = }}{{\rm{C}}_{{\rm{K-mer}}}}{\rm{ \times L}}\left( {{\rm{L - K = 1}}} \right){\rm{;}}}\\ & {{\rm{G = }}{{\rm{N}}_{{\rm{K - mer}}}}{\rm{/}}{{\rm{C}}_{{\rm{K-mer}}}}{\rm{ = }}{{\rm{N}}_{{\rm{base}}}}{\rm{/}}{{\rm{C}}_{{\rm{base}}}}} \end{split} $
式中:Nbase和NK-mer为序列的碱基总数和K-mer数,Cbase和CK-mer为覆盖碱基的期望深度和K-mer期望覆盖深度。
对预估的基因组大小进行修正,将K-mer深度为1的情况认为是错误情况,计算错误率,并用于修正基因组大小,修正公式为
$ {{\rm{G}}_{{\rm{revised}}}}{\rm{ = Genome \times }}{\rm{(1 - E)}} $
式中:Grevised为修正后的基因组大小,E为测序错误率。
通过K-mer数学分析模型,基因组杂合率公式为:
$ X = \frac{{a_{1/2} \times n_K/(2\times K)}}{{n_K - a_{1/2} \times n_K/2}} = \frac{{a_{1/2}}}{{K\left( {2 - a_{1/2}} \right)}} $
式中:a1/2为杂合K-mer种类数的百分比,nK为所有K-mer的种类数。
另外,计算标准泊松分布和实际数据曲线峰值后的面积差值,可得到重复序列百分比,在这里我们计算纯合峰深度1.8倍后面的K-mer个数所占的比例来估计重复序列比例。
-
由于西伯利亚白刺基因组重复序列较多,我们选择K-mer=41将打断的DNA序列拼接组装到Scaffold,通过reads之间的overlap关系构建de Bruiji图并对其简化,在重复区域边界位置进行剪切,得到contig序列,再根据大片段数据的Pair-end关系,构建Scaffold序列,最后用reads对Scaffold的gap区域进行填补,完成组装过程,具体配置参数为
pregraph : -K 41 -R -d 1
-K kmer: K value in kmer
-R (optional): unsolve repeats by reads (default no)
-d KmerFreqCutoff(optional): delete kmers with frequency no larger than (default 0)
contig : -D 1 -M 1 -R
-D EdgeCovCutoff(optional): delete edges with coverage no largert than (default 1)
-M mergeLevel (default 1,min 0, max 3): the strength of merging similar sequences during contiging
-R solve_repeats (optional): solve repeats by read paths(default: no)
map : -K 41
-K kmer (default: the same as in pregraph): k value in kmer
scaff : -F 1 -L 43
-F (optional) fill gaps in scaffold. (default 0; 1:normally; -1:only fill nonrepeat gap; 2:radically)
-L minLen : shortest contig (minus K value) for scaffolding
再根据组装结果统计其contig分布情况,统计测序长度大于500 bp的测序深度和GC含量并做GC含量分布图。
-
运行MISA脚本(pgrc.ipk-gatersleben.de/misa)对过滤后数据SSR位点鉴定并统计其类型、数量。筛选标准为单核苷酸SSR位点≥16次,双核苷酸SSR位点≥6次,三四核苷酸SSR位点≥5次。
-
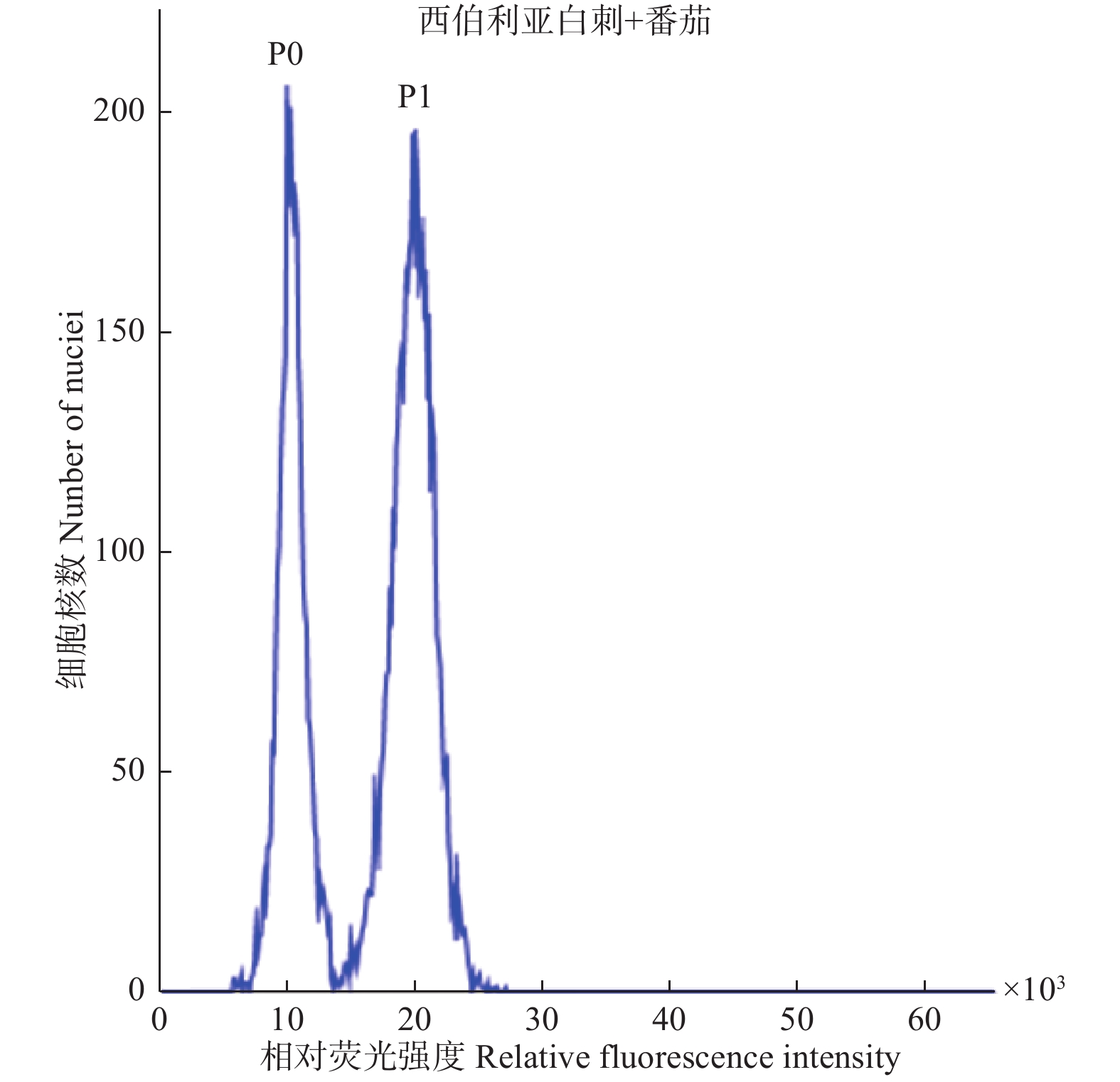
将西伯利亚白刺和番茄的叶片混合解离液放入流式细胞运行并在480 nm波长下检测其荧光吸收强度(图1),其中,P0为西伯利亚白刺的吸收峰,P1为番茄的吸收峰,番茄参考2C值为1.96 pg,实验重复3次。将平均值代入C值计算公式得出:2C西伯利亚白刺=2C番茄×(G0/G1西伯利亚白刺)/(G0/G1番茄)=1.96 pg×0.534,得西伯利亚白刺C值大小为523.4 Mbp。
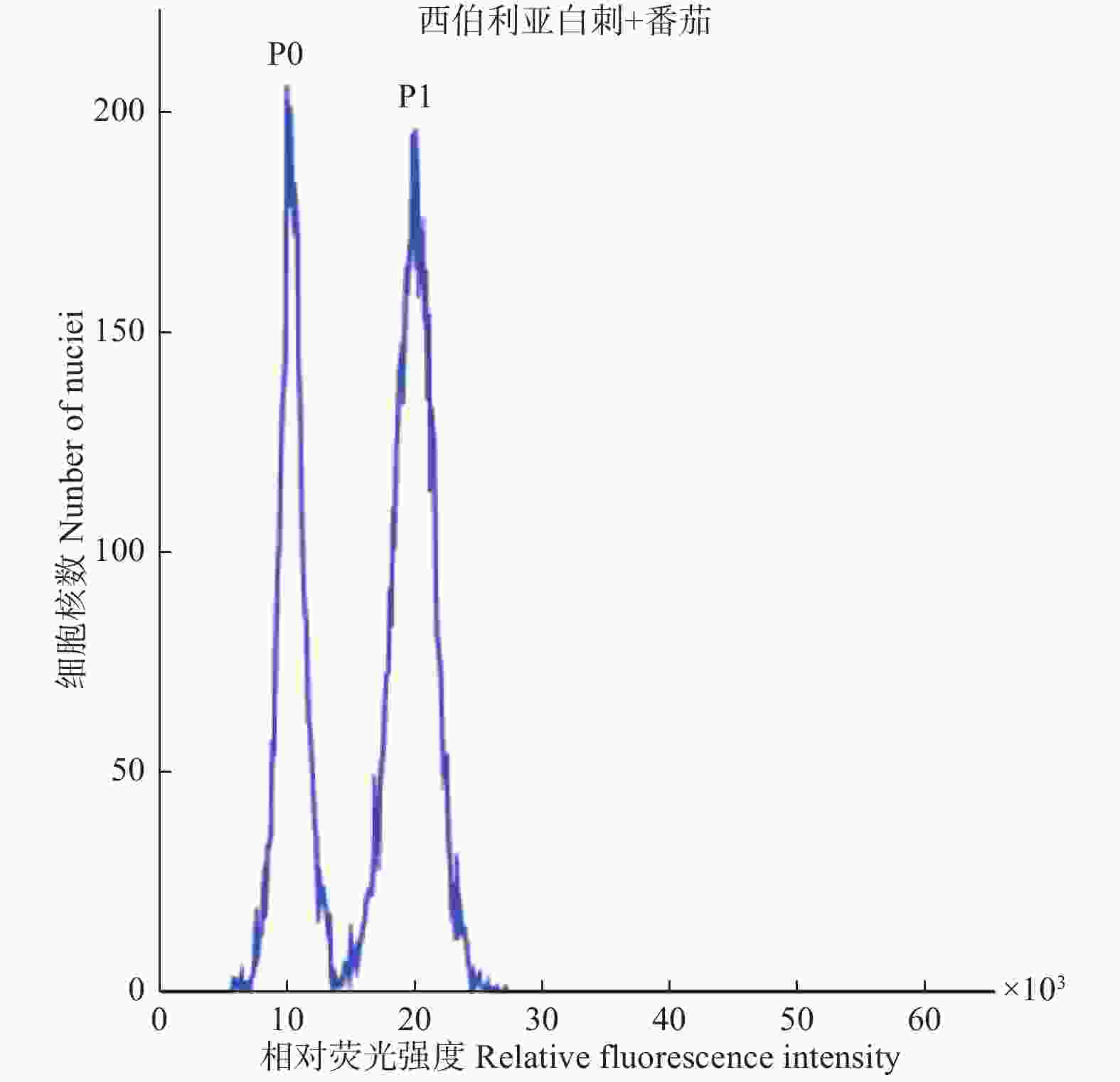
图 1 流式细胞测定结果
Figure 1. Flow cytometry results
-
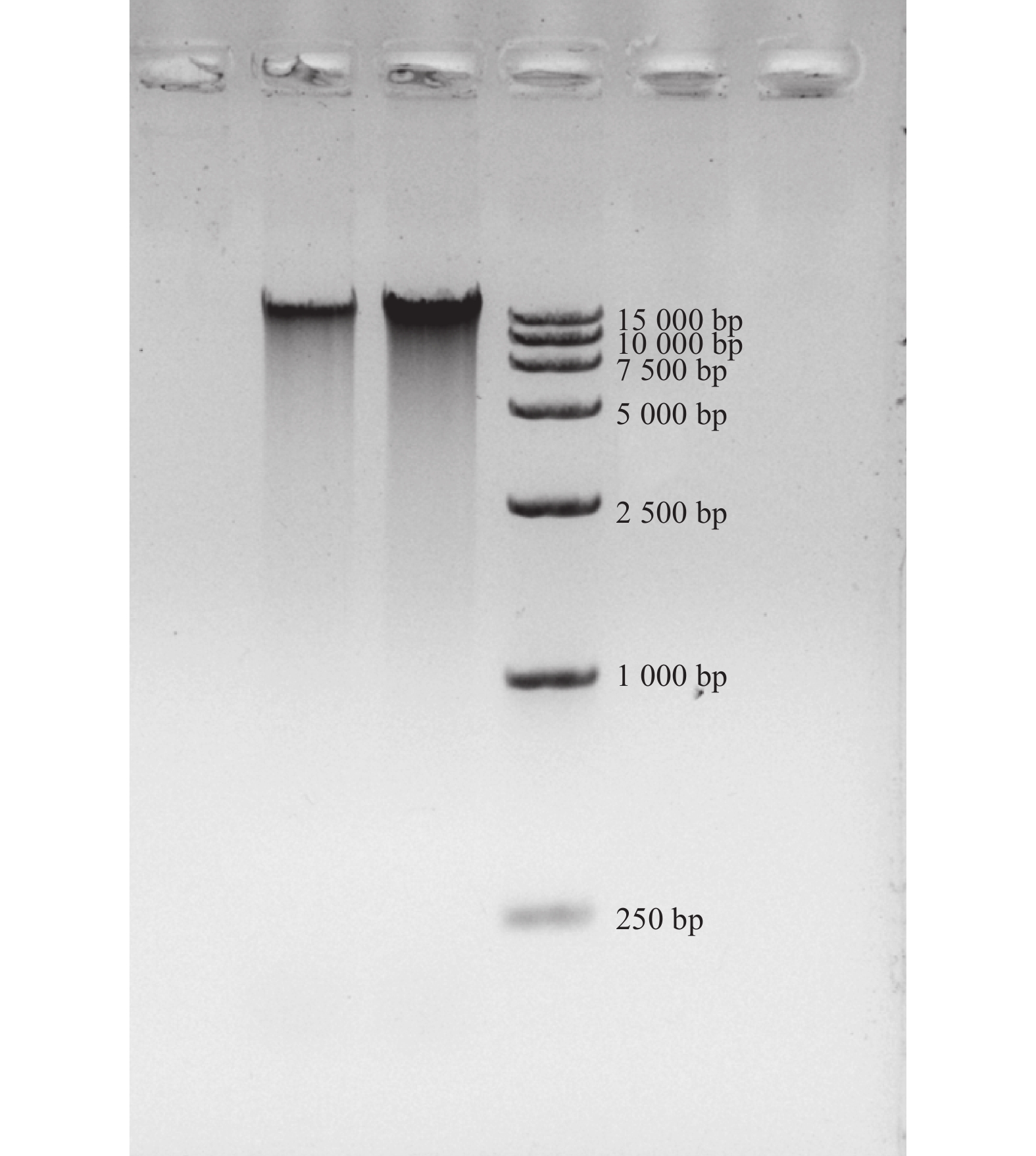
取1 μL DNA样品于分光光度计的检测,结果显示OD260/OD280为1.89,浓度为206.9 ng·μL−1。再利用1%琼脂糖凝胶电泳检测其条带完整性,图2 表明:电泳条带单一,无明显杂带。综合二者推测,此DNA完整度较高,可用于下游实验。
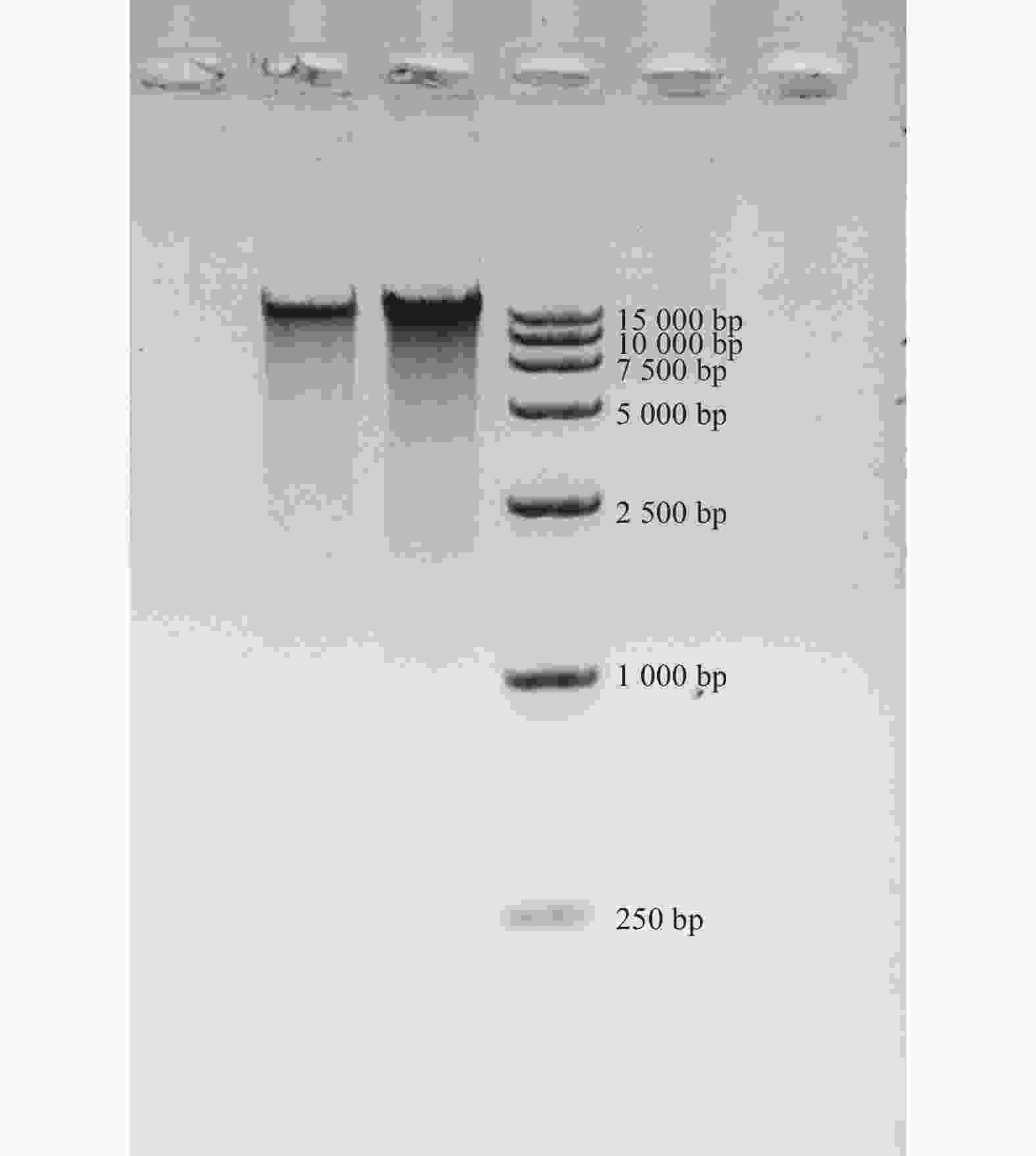
图 2 DNA琼脂糖凝胶电泳图
Figure 2. DNA agarose gel electrophoresis
-
过滤掉无效或低质量的reads数据,再经图像识别、去污染等步骤,得出最终的测序结果(表1):其中,测序的总reads数为212 852 294个,测序的总数据大小为63 855.69 Mbp,按照536.16 Mbp的预估基因组大小得出本次测序深度为119.09×,测序的错误率为0.04%,Q20的含量为95.59%,Q30的含量为89.33%,GC含量为36.78%。
表 1 测序结果统计
Table 1. Sequencing results statistics
reads数量
Raw paired
reads数据产量
Raw base/
Mbp测序错误率
Error rate/%Q20含量
Q20
Content/%Q30含量
Q30
Content/%GC含量
GC
Content%212 852 294 63 855.69 0.04 95.59 89.33 36.78 -
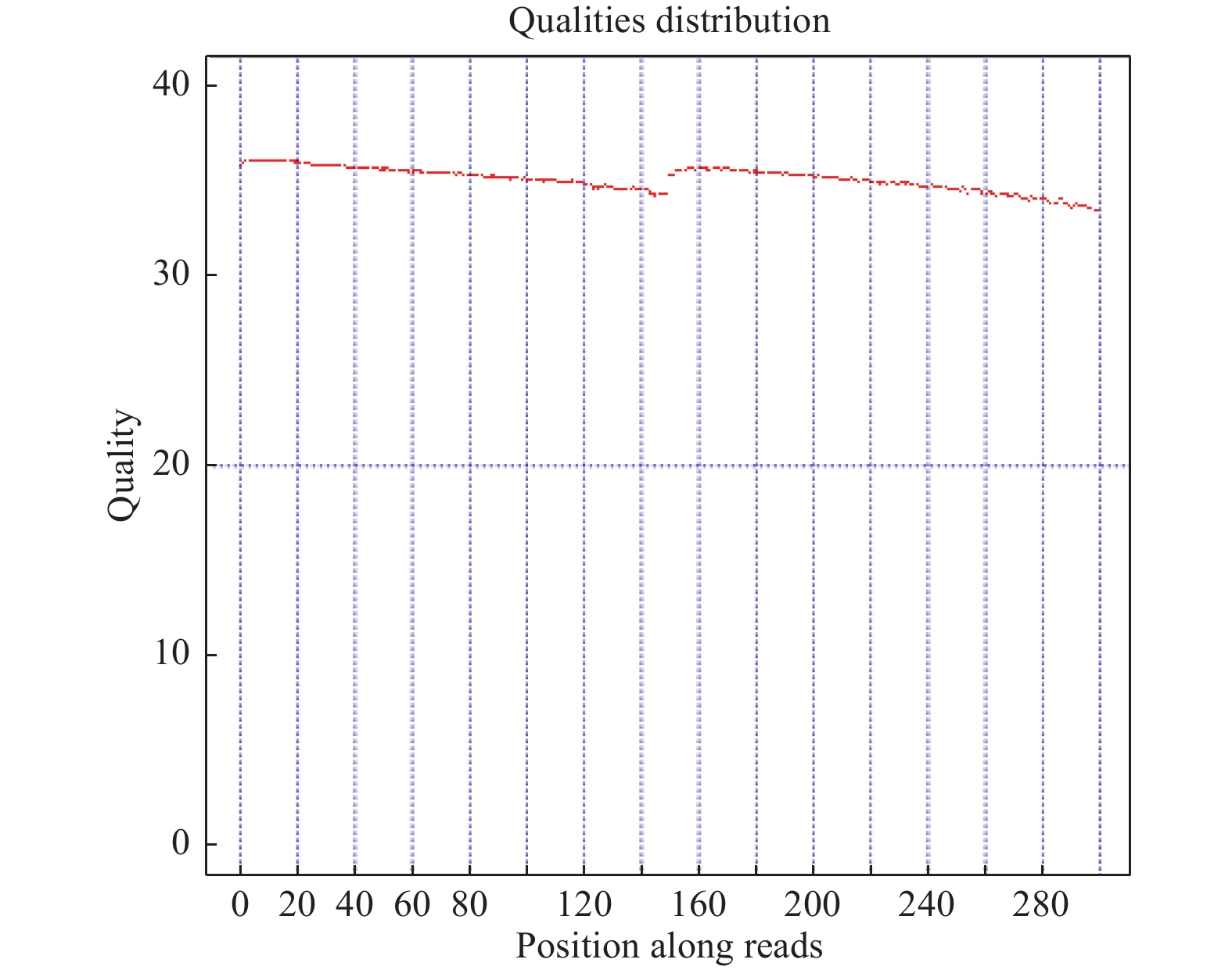
测序数据的质量主要分布在Q30(≥80%)以上,这样才能保证后续分析的正常进行,如图3所示,实验Q30含量为89.33%满足后续分析要求。
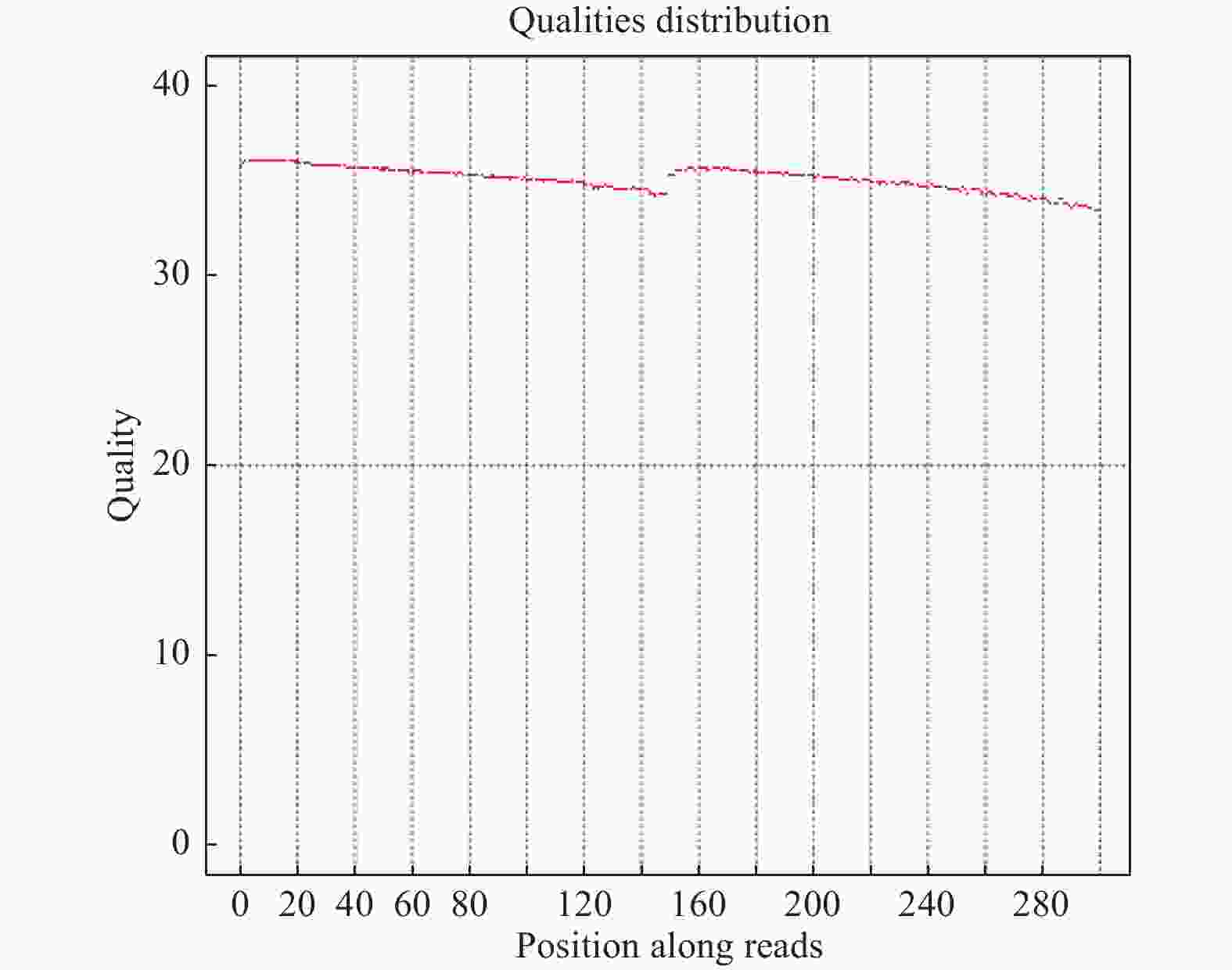
图 3 数据质量分布
Figure 3. Data quality distribution
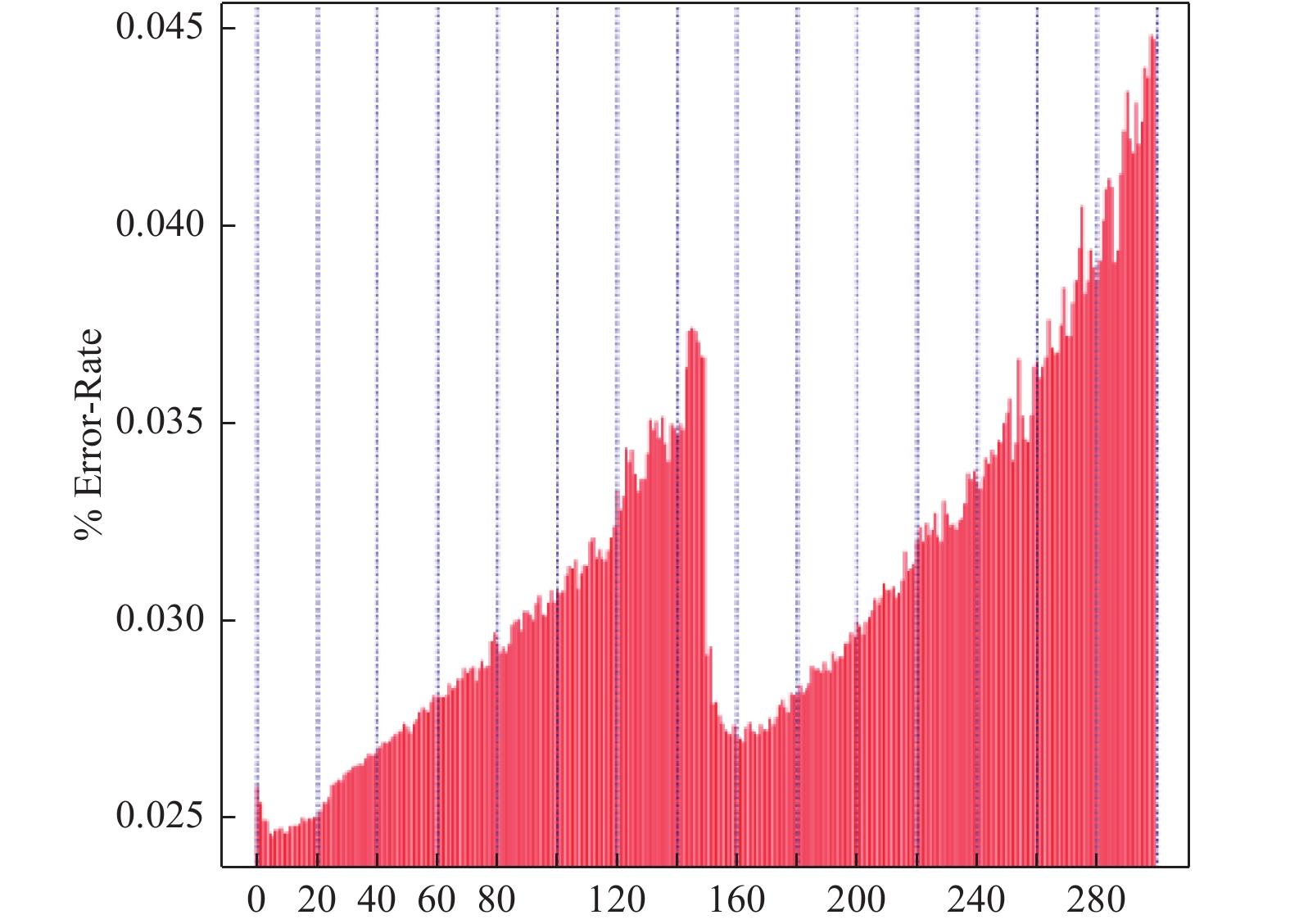
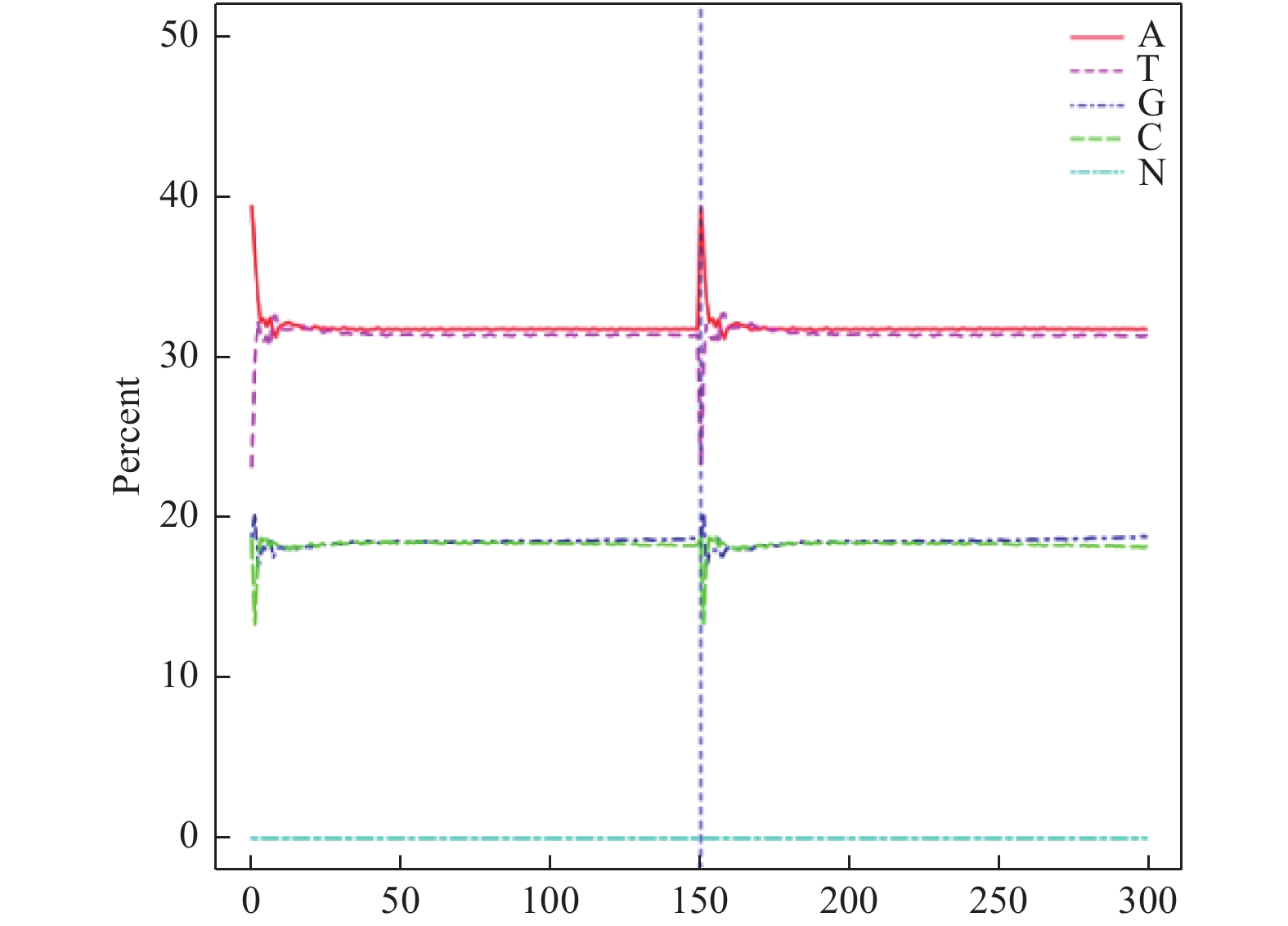
此外,测序错误率也影响测序结果的准确性,对于下游分析至关重要,本实验2个reads的测序错误率均低于1%(图4),表明本次测序错误率控制良好。为进一步保证测序结果的可信性,还需对本次测序的碱基含量分布进行分析。GC含量分布检查用于检测有无AT、GC分离现象,理论上G和C含量以及A和T含量在每个测序循环上应分别相等,且整个测序过程中稳定不变,呈水平线。由于DNA模板扩增偏差等原因使测序前几个碱基测序质量值较低,发生小幅度波动,属于正常情况。本实验中(图5)测序的G和C的含量和A和T的含量接近也保证了测序的可信度。
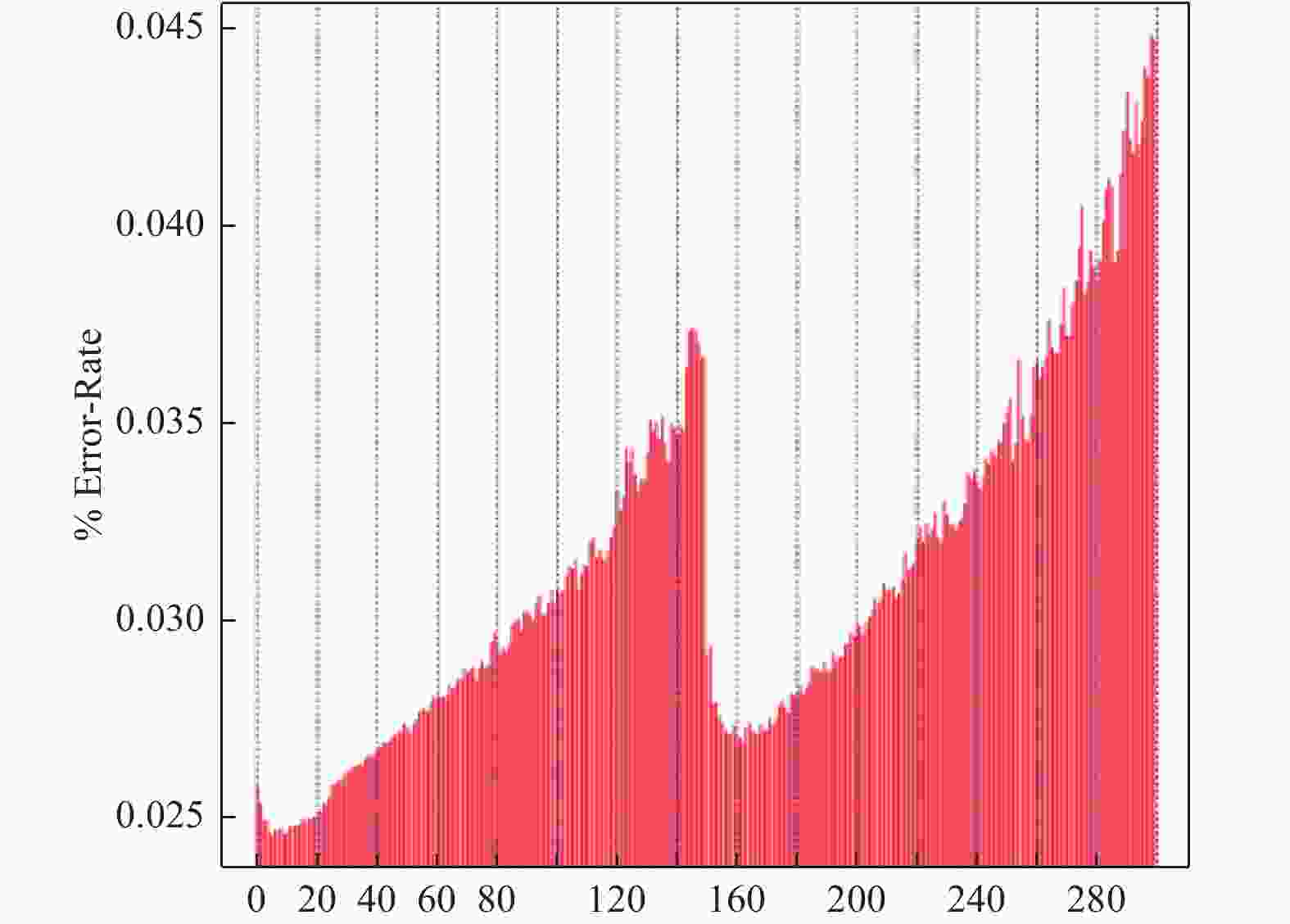
图 4 测序错误率分布
Figure 4. Sequencing error rate distribution
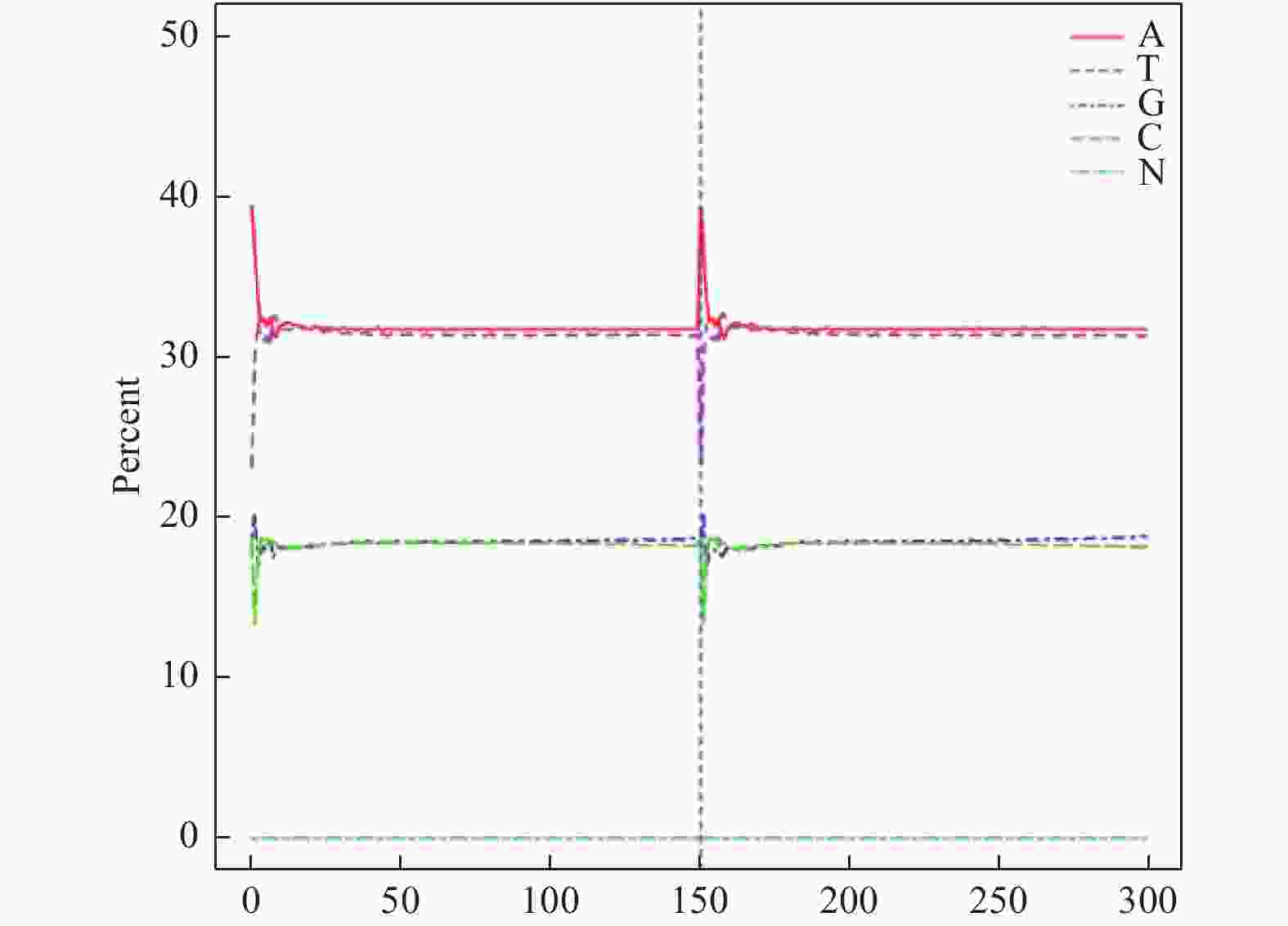
图 5 GC含量分布图
Figure 5. GC content distribution map
-
利用K-mer分析法对西伯利亚白刺基因组大小进行估计,根据测序结果(表2、图6)发现:当K-mer深度为89×时存在明显的主峰,由K-mer相关公式计算得到的基因组大小为536.16 Mbp,并通过后续基因修正得修正后基因组大小为526.30 Mbp; 而在主峰前横坐标二分之一处出现次峰。一般当目标序列存在杂合现象时,存在杂合位点的K-mer被分成2份,频率变成原频率的1/2,因此,此峰为杂合峰,并统计得出西伯利亚白刺基因组杂合率为0.90%,杂合率较高,属于复杂基因组。此外,在约为主峰2倍depth的地方存在次峰,并有明显的拖带现象,该片段出现的期望值是大部分的2倍,这些片段为重复片段,由相关统计结果得重复序列数占总序列数的55.39%。
表 2 K-mer=17分析所得各项数据
Table 2. K-mer=17 analysis of the data
数据名称Data name 数据大小Data size K-mer数/bp 47 718 560 459 修正前基因组大小/Mbp 536.16 修正基因组大小/Mbp 526.30 杂合率/% 0.90 重复率/% 55.39 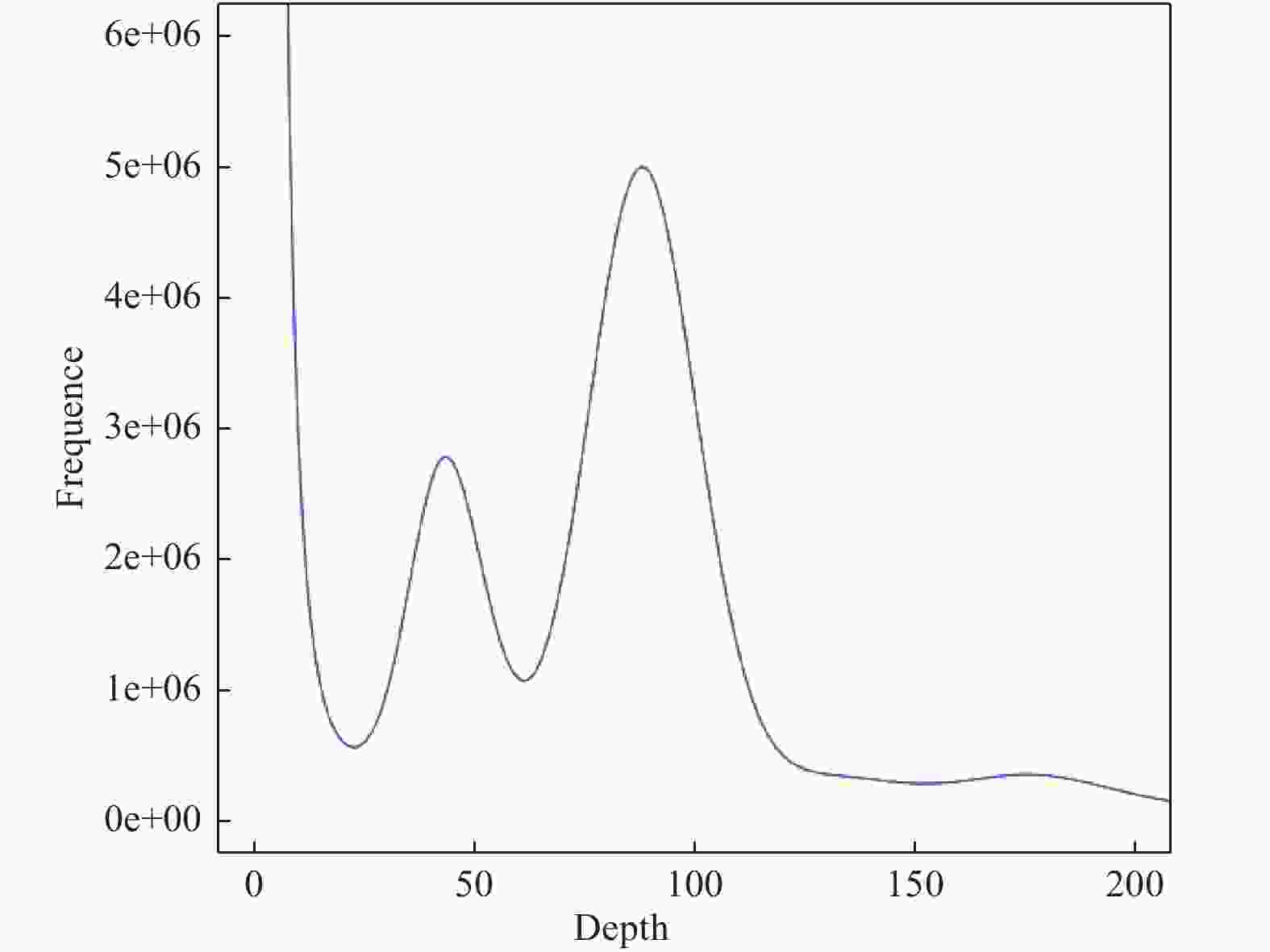
图 6 K-mer=17 Depth和K-mer种类数频率分布图
Figure 6. K-mer=17 Depth and K-mer species frequency distribution
-
运用Soapdenovo软件拼接上述测序数据,并对数据进行纠错,构建contig、scaffold等优化过程,得到初步的基因组组装信息(表3):针对组装好的长度大于等于100 bp的scaffold内部contig进行统计,得N50长度为1 076 bp,N90为147 bp,组装得到最长的序列长度为45 660 bp,组装的contig总数量为917 423个,总长度为424 458 883 bp。进一步将所有文库测序得到的reads比对回初步得到的contigs,利用reads之间的连接关系和插入片段大小信息,过滤掉长度<100 bp的contig序列,最终将contigs组装成scaffolds,结果显示:N50的长度的1 889 bp,N90为189 bp,最长序列长度为89 063 bp,组装总量为717 232个,总长度为443 258 576 bp。
表 3 基因组组装结果统计
Table 3. Genomic assembly results statistics
数据类型Types of data 拼接序列Spliced sequence Contig Scaffold Total_length/bp 424 458 553 443 258 576 N90_length/bp 147 189 Max_length/bp 45 660 89 063 N50_length/bp 1 076 1 889 Total_number/个 917 423 717 232 -
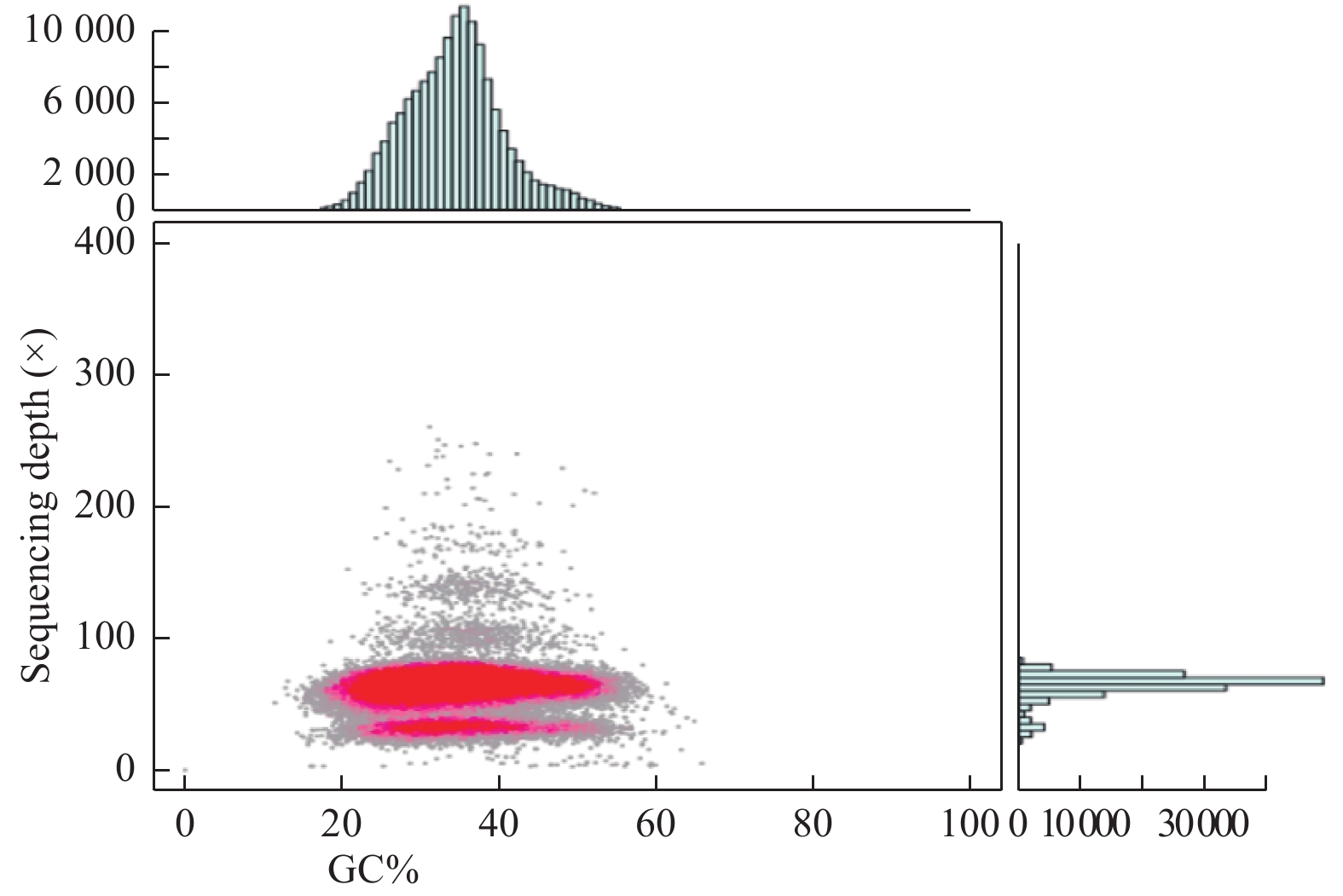
GC含量是反映植物基因组成的重要指标之一,GC含量深度分析图用于检测测序是否存在GC分布偏向,样品是否存在细菌的污染等。由图7可得:西伯利亚白刺基因组测序没有明显的GC偏向。图中有2处GC聚集处,为了确认低测序深度区域是否为细菌污染造成,将低测序深度序列比对到NCBI核苷酸数据库,并没有细菌序列被比对上,说明样品没有被细菌污染,推测这是由于西伯利亚白刺基因组高杂合度所造成的。由于在组装过程中同源染色体上杂合部位只能被识别出一半,导致此部位的GC含量分布在低测序深度区域。
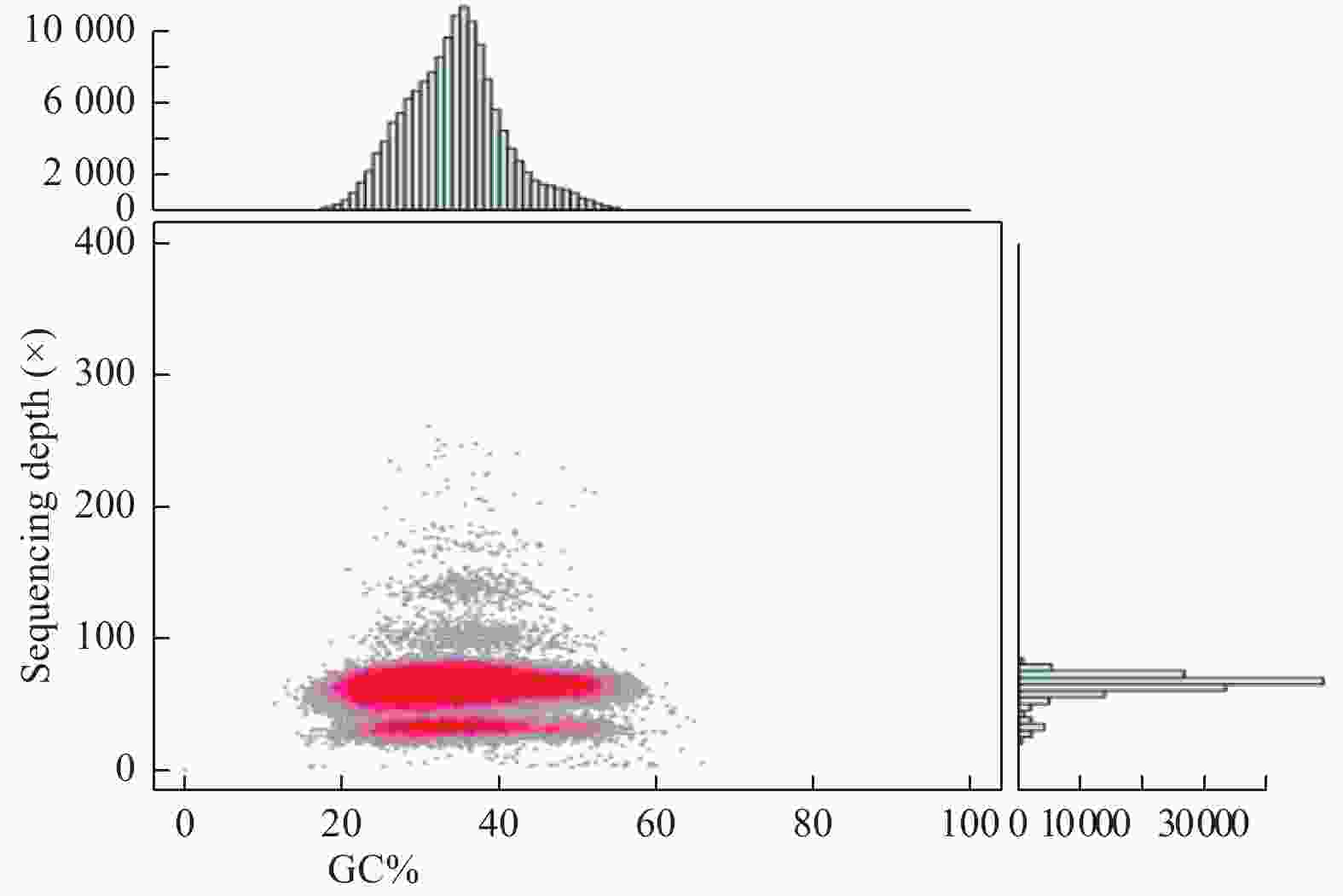
图 7 GC含量与测序深度关联分析统计图
Figure 7. GC content and sequencing depth correlation analysis
-
由MISA脚本分析西伯利亚白刺基因组数据并统计(表4),共搜寻到521 125个SSR位点,其中,单核苷酸位点出现比例最高,达342 883个,占总SSR位点的65.80%;二核苷酸位点146 312个,占比28.06%;三核苷酸位点26 133个,占5.02%;四个及以上核苷酸位点8 678个,占1.67%。所以,单核苷酸重复是西伯利亚白刺主要的SSR重复位点,同时单核苷酸重复中A/T占比最多,达到了63.94%。
表 4 西伯利亚白刺SSR位点统计
Table 4. SSR locus statistics of N. sibirica
类型Type 结构structure 数目Number 所占比例Proportion/% 单碱基 A/T 333 188 63.94 G/C 9 695 1.86 双碱基 AC/GT 10 978 2.10 AG/CT 14 095 2.70 AT/AT 118 038 22.65 GG/CG 3 201 0.61 AAC/GTT 946 0.18 三碱基 AAG/CTT 4 311 0.83 AAT/ATT 16 521 3.17 ACC/GGT 1 601 0.31 ACG/CGT 87 0.02 ACT/AGT 121 0.02 AGC/CTG 452 0.09 AGG/CCT 653 0.13 ATC/ATG 1 259 0.24 CCG/CGG 182 0.03 ≥四碱基 8 678 1.67 合计 521 125 100 -
基因组大小是指生物单倍体染色体中DNA的含量,也称为C值[18]。目前为止已有数千种动植物的C值被检测并收录入相应的动植物C值库[19-20]。DNA的C值是生物体重要的基因特征,是种群分类的证据之一,也是开展各项基因工作的基础。了解基因组大小对于推测物种的演化趋势、进化地位、种属间进化关系、生物进化分类等具有深远的意义。
基因组大小预测常使用流式细胞术[21]、Feulgen图像分析法[22]、全基因组survey调查[23]等方法。流式细胞术通过比较待测植物和内标植物细胞悬液荧光吸收峰比值,根据公式由内标植物的基因组来计算待测植物基因组的大小,是一种快速、便捷的基因组预估的方法,在测定动植物体的基因组大小方面均有较广的应用。
全基因组survey测序是基于小片段文库的低深度从头测序,通过对原始数据进行图像识别,去污染、去接头等步骤,再进行K-mer分析,Soapdenovo软件组装继而完成整个分析过程,可对基因组的大小、GC含量、杂合率以及重复序列的含量等重要的基因组特征信息进行分析,相比于流式细胞仪、Feulgen图像分析法等基因组大小预测方法更能切合所测生物体基因组特征,是一种更精确的分析未知基因组特征的途径[24-26]。
西伯利亚白刺基因组GC含量为36.78%,没有明显的过高或过低的情况[27],对NGS测序准确性影响较小;而其杂合率为0.9%,基因组重复序列比例达55.39%,属于高杂合基因组。推测可能是由于西伯利亚白刺在地理分布上较广,生态条件悬殊、植物形态变化也较大有关[28]。
一般来说,基因组杂合度越大,重复片段越多,该物种的组装难度就越大。西伯利亚白刺属于高杂合基因组植物,而同为高杂合基因组的胡杨利用全基因组鸟枪法结合Fosmid拼装策略获得了精度较高的基因组图谱[29]具有一定参考意义,如果使用二代测序Platanus组装软件[30]可能更适合于西伯利亚白刺基因组的拼装。随着近年来测序成本的下降和3代测序技术的普及,二代llumina搭配三代Pacbio辅以Hi-C技术的方案将会是西伯利亚白刺全基因组测序更好的选择,更有利于获得高质量的全基因组图谱。
-
本实验测得西伯利亚白刺基因组大小为536.16 Mbp,修正后为526.30 Mbp,杂合率为0.90%,重复序列比例为55.39%;西伯利亚白刺Contig N50为1 076 bp,总长为424 458 553 bp,Scaffold N50为1 889 bp,总长为443 258 576 bp。西伯利亚白刺有521 125个SSR位点,其中单核苷酸位点有342 883个,二核苷酸位点有146 312个,三核苷酸位点有26 133个,四个及以上为8 678个,单核苷酸为其主要的SSR特征。
西伯利亚白刺基因组信息初探
A study on Nitraria sibirica Pall Genome
-
摘要:
目的 为了解西伯利亚白刺(Nitraria sibirica Pall)基本基因组信息,如基因组大小、复杂程度等特征。 方法 (1)以西伯利亚白刺的幼苗嫩叶为材料,以番茄为标定,用解离液mG解离叶片后,经流式细胞仪检测细胞悬液,收集细胞并计算西伯利亚白刺的C值。(2)通过构建小片段文库,进行Illumina HiSeq测序并分析测序结果,预估其基因组大小、GC含量、杂合率、重复序列比例等,使用MISA脚本对过滤后数据进行SSR位点分析。 结果 西伯利亚白刺预估全基因组大小为526.30 Mbp,基因组GC含量为36.78%,基因组杂合率为0.90%,重复序列比例为55.39%。对测序数据进行SSR位点标记,共得到521 125个SSR位点,其中,单核苷酸重复为342 883个,占总SSR位点的65.80%。 结论 由各项分析指标推测西伯利亚白刺为复杂基因组,可以采用二代Illumina与三代PacBio测序辅以Hi-C技术联合分析,有利于西伯利亚白刺基因组的组装及基因图谱的获取。 Abstract:Objective This study aims at estimating the size, complexity and other characteristics of Nitraria sibirica Pall genome. Method Taking Solanum lycopersicum as DNA reference standard, the young leaves of N. sibirica were used as test material and mG as separate buffer to establish the N. sibirica genomic C-value by flow cytometry (FCM) method. A small fragment library was established for Illumina HiSeq sequencing. The genome size, heterozygosity, GC content and repetitive sequence ratio were studied and MISA script was used to analyze the SSR data. Result The size of N. sibirica genome was 526.30 Mbp, the heterozygosity rate was 0.90%, the GC content ratio was 36.78% and the repeat sequence ratio was 55.39%. Using the assembled genome data for SSR analysis obtained 521 125 loci, of which mononucleotide repeat were 342 883 loci, accounting for 65.80% of the total SSRs. Conclusion It is predicted that the N. sibirica genome has a complex nature and can be analyzed via Illumina combined to PacBio and Hi-C technologies in order to facilitate the assemble of N. sibirica genome and the acquisition of genetic maps. -
Key words:
- Nitraria sibirica Pall
- / Flow cytometry
- / genome size
- / GC content
- / SSR molecular marker
-
图 6 K-mer=17 Depth和K-mer种类数频率分布图
Figure 6. K-mer=17 Depth and K-mer species frequency distribution
表 1 测序结果统计
Table 1. Sequencing results statistics
reads数量
Raw paired
reads数据产量
Raw base/
Mbp测序错误率
Error rate/%Q20含量
Q20
Content/%Q30含量
Q30
Content/%GC含量
GC
Content%212 852 294 63 855.69 0.04 95.59 89.33 36.78  下载: 导出CSV
下载: 导出CSV
表 2 K-mer=17分析所得各项数据
Table 2. K-mer=17 analysis of the data
数据名称Data name 数据大小Data size K-mer数/bp 47 718 560 459 修正前基因组大小/Mbp 536.16 修正基因组大小/Mbp 526.30 杂合率/% 0.90 重复率/% 55.39
下载: 导出CSV
表 3 基因组组装结果统计
Table 3. Genomic assembly results statistics
数据类型Types of data 拼接序列Spliced sequence Contig Scaffold Total_length/bp 424 458 553 443 258 576 N90_length/bp 147 189 Max_length/bp 45 660 89 063 N50_length/bp 1 076 1 889 Total_number/个 917 423 717 232
下载: 导出CSV
表 4 西伯利亚白刺SSR位点统计
Table 4. SSR locus statistics of N. sibirica
类型Type 结构structure 数目Number 所占比例Proportion/% 单碱基 A/T 333 188 63.94 G/C 9 695 1.86 双碱基 AC/GT 10 978 2.10 AG/CT 14 095 2.70 AT/AT 118 038 22.65 GG/CG 3 201 0.61 AAC/GTT 946 0.18 三碱基 AAG/CTT 4 311 0.83 AAT/ATT 16 521 3.17 ACC/GGT 1 601 0.31 ACG/CGT 87 0.02 ACT/AGT 121 0.02 AGC/CTG 452 0.09 AGG/CCT 653 0.13 ATC/ATG 1 259 0.24 CCG/CGG 182 0.03 ≥四碱基 8 678 1.67 合计 521 125 100
下载: 导出CSV
-
[1] 赵可夫, 范 海, 江行玉, 等. 盐生植物在盐渍土壤改良中的作用[J]. 应用与环境生物学报, 2002, 8(1):31-35. doi: 10.3321/j.issn:1006-687X.2002.01.006 [2] 玉苏甫•买买提, 阿布都克里木•热依木, 阿丝叶•阿不都力米提. 西伯利亚白刺不同部位的氨基酸测定分析[J]. 中国农学通报, 2016, 32(22):37-41. doi: 10.11924/j.issn.1000-6850.casb16020047 [3] 耿庆祯, 都恕兴, 尚艳霞, 等. 西伯利亚白刺与唐古特白刺果实营养成分比较分析[J]. 食品科技, 2008, 33(3):101-104. doi: 10.3969/j.issn.1005-9989.2008.03.029 [4] 高 航, 索有瑞. 柴达木盆地西伯利亚白刺和唐古特白刺的氨基酸含量及其营养评价[J]. 氨基酸和生物资源, 2002, 24(4):4-7. doi: 10.3969/j.issn.1006-8376.2002.04.002 [5] 王晨霞, 陈贵林. 西伯利亚白刺的组织培养与快速繁殖[J]. 植物生理学通讯, 2007, 43(6):1143-1144. [6] 玉苏甫•买买提, 阿丝叶•阿不都力米提, 尤努斯•居玛. 于田县西伯利亚白刺播种育苗技术研究[J]. 北方园艺, 2016(19):71-74. [7] Banaev E V, Vysochina G I, Kukushkina T A, et al. Variability in the content of biologically active substances in the leaves ofNitraria sibirica Pall. (Nitrariaceae)[J]. Contemporary Problems of Ecology, 2014, 7(1): 90-96. doi: 10.1134/S1995425514010028 [8] Mamat Y, Reyim A, Abdulimit A. Detection and analysis of amino acids in differentparts of Nitraria sibirica[J]. Chinese Agricultural Science Bulletin, 2016. [9] 李焕勇, 唐晓倩, 杨秀艳, 等. NaCl处理对西伯利亚白刺幼苗中矿质元素含量的影响[J]. 植物生理学报, 2017, 53(12):2125-2136. [10] 陈志强, 李庆贱, 时瑞亭, 等. 苏打盐碱胁迫对西伯利亚白刺光合和生长的影响[J]. 北京林业大学学报, 2011, 33(3):31-37. [11] Wang L, Ma Y K, Li N N, et al. Isolation and characterization of a tonoplast Na+/H+ antiporter from the halophyte Nitraria sibirica[J]. Biologia Plantarum, 2016, 60(1): 113-122. doi: 10.1007/s10535-015-0560-8 [12] Li H, Tang X, Yang X, et al. De novo transcriptome characterization, gene expression profiling and ionic responses of Nitraria sibirica Pall. under salt stress[J]. FORESTS, 2017, 8(2116): 211. [13] 陈 勇, 柳亦松, 曾建国. 植物基因组测序的研究进展[J]. 生命科学研究, 2014, 18(1):66-74. [14] Goff S A, Ricke D, Lan T H, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. Japonica)[J]. Science, 2002, 296(5565): 92-100. doi: 10.1126/science.1068275 [15] Gregory T R. Animal genome size database[DB]. Noncoding DNA, 2001. [16] 唐 其, 马小军, 莫长明, 等. 罗汉果全基因组Survey分析[J]. 广西植物, 2015, 35(6):786-791. [17] 张 伟, 王 瑞, 杨晓野, 等. 捕食性真菌Duddingtonia Flagrans基因组DNA的提取及基因组Survey分析[J]. 中国兽医学报, 2017(11):2090-2094. [18] Shi M J, Cheng Y Y, Zhang W T, et al. The evolutionary mechanism of genome size[J]. Chinese Science Bulletin, 2016. [19] Ryan G T, Nicol J A, Heidi T, et al. Eukaryotic genome size databases[J]. Nucleic Acids Research, 2007, 35(Database issue): D332-D338. [20] Sònia G, Leitch I J, Alba A R, et al. Recent updates and developments to plant genome size databases[J]. Nucleic Acids Research, 2014, 42(Database issue): D1159-D1166. [21] Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry[J]. Nature Protocols, 2007, 2(9): 2233-2244. doi: 10.1038/nprot.2007.310 [22] Jeffery N W, Gregory T R. Genome Size Estimates for Crustaceans Using Feulgen Image Analysis Densitometry of Ethanol-Preserved Tissues[J]. Cytometry Part A the Journal of the International Society for Analytical Cytology, 2015, 85(10): 862-868. [23] 王 雪, 周佳熠, 孙会改, 等. 新疆沙冬青基因组调查测序与基因组大小预测[J]. 植物遗传资源学报, 2018, 19(1):143-149. [24] 孟金涛, 苑建蕊, 魏彦杰, 等. 基于DeBruijn图的DeNovo序列组装软件性能分析[J]. 科研信息化技术与应用, 2013, 29(5): 39-41. [25] Li R, Fan W, Tian G, et al. The sequence and de novo assembly of the giant panda genome[J]. Nature, 2010. [26] Miller J R, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data[J]. Genomics, 2010, 95(6): 315-327. doi: 10.1016/j.ygeno.2010.03.001 [27] Aird D, Ross M G, Chen W S, et al. Analyzing and minimizing PCR amplification bias in illumina sequencing libraries[J]. Genome Biology, 2011, 12(2): R18. doi: 10.1186/gb-2011-12-2-r18 [28] Zhang M, Temirbayeva K, Sanderson S C, et al. Young Dispersal of Xerophil Nitraria Lineages in Intercontinental Disjunctions of the Old World[J]. Scientific reports, 2015, 5(1): 13840. doi: 10.1038/srep13840 [29] Ma T, Wang J, Zhou G, et al. Genomic insights into salt adaptation in a desert poplar[J]. Nature Communications, 2013, 4(4): 2797. [30] Cong Q, Borek D, Otwinowski Z, et al. Tiger swallowtail genome reveals mechanisms for speciation and caterpillar chemical defense[J]. Cell Reports, 2015, 10(6): 910-919. doi: 10.1016/j.celrep.2015.01.026 -
 点击查看大图
点击查看大图
图(7) / 表(4)
计量
- 文章访问数: 4526
- HTML全文浏览量: 2710
- PDF下载量: 45
- 被引次数: 0



