


-
杉木(Cunninghamia lanceolata (Lamb.) Hook)是我国南方林区重要的速生用材造林树种,经过40多年的遗传改良,选育了一大批速生优质的杉木新品种应用于生产中。杂交育种是杉木遗传改良的主要途径[1],主要通过选择遗传互补的优良亲本进行交配,再选择出速生优质的杂交新品种。在杉木杂交育种研究中,生长性状的杂种优势现象已有报道[2],主要从数量遗传学或表观遗传学等方面进行了探讨[2-4]。
随着分子生物学技术不断发展,利用分子标记、RNA差异显示、基因芯片等技术对杂种优势已有所研究,如Birchler认为基因差异表达模式与杂种F1的表型存在一定的联系,杂种的特定性状可能是父本、母本特定位点的等位基因共同表达的结果[5-6]。Li等[7]利用基因芯片技术对落叶松(Larix spp.)的杂种优势研究表明,杂种F1与亲本间有54个差异显著基因为非加性表达模式,且这些基因参与生理过程、应激反应及淀粉和蔗糖代谢等多种生化途径。丁昌俊等[4]运用转录组测序法研究了不同生长势的美洲黑杨(Populus deltoides Marsh)转录组的差异,获得了大量差异基因,发现杨树杂种优势的形成可能是由于相关基因的显著表达,调节光合作用、物质代谢吸收等与生长紧密联系的代谢活动,进而促进了杂种生长优势。
不少学者借助转录组测序或RNA-seq高通量测序技术, 研究了杉木纤维性状发育的形成机制和木材形层成活动的机理[8-12],但杉木生长性状杂种优势产生的分子机理研究尚未见报道。本研究利用无参转录组测序技术,在基因差异研究的基础上,以基因差异表达分析为切入点,以杉木1个优良组合的超亲、低亲两组杂交子代及其亲本为研究材料,展开了杂种子代和亲本两两间的比较分析,以期揭示杉木生长性状杂种优势的分子机理,获得具有重要生物学功能的基因,为杉木杂交育种提供科学依据。
HTML
-
研究材料为已选育出的杉木优良杂交组合(龙15×1339)及其亲本。1996年春,对前期试验中表现突出的杉木杂交组合,在浙江遂昌杉木种子园进行了重复制种,1997年完成杂交子代育苗。同年,将浙江遂昌多系种子园几个小区改建成龙15×1339等组合的双系种子园。1997年嫁接成园。本研究中使用的亲本和杂种(龙15×1339)不仅林龄相同,且都达到成熟阶段。
2017年6月21日,对杉木双系种子园中龙15和1339及同龄区域化试验林中的龙15×1339子代取样,叶样来自样树顶部当年生的嫩枝叶,3个生物学重复,龙15(P1)3个样株的编号为P1-1、P1-2和P1-3,1339(P2)3个样株的编号为P2-1、P2-2和P2-3。由于杂交组合内子代会发生分离,杂种子代的取样参照丁昌俊等[4]的方法。对试验林内该组合抽取超亲子代3株:HF1-1、HF1-2、HF1-3,低亲子代3株:LF2-1、LF2-2、LF2-3,构成4个样本组HF1、LF2、P1和P2,进行测序、序列组装、功能注释,最后亲本与子代共4个样本组,两两比较,总共6个比较组进行处理与分析。杉木龙15×1339子代表现和超亲杂种优势见表 1。
杂交组合
Cross combinations材积
Volume/dm3树高
Height/m胸径
DBH/cm龙15×1339
Long 15×133919.69
(20.87%)5.75
(7.48%)8.35
(8.58%)龙15×闽33
Long 15×Min3318.51
(13.63%)5.46
(2.06%)8.21
(6.76%)龙15半同胞
Long 15 family of half siblings16.29 5.35 7.69 注:括号中的数值是超亲优势。
Note: The value in parentheses is transgressive advantage.Table 1. The superior advantages of the superior combinations Long 15×1339 of Zhejiang Suichang
取样时超亲子代HF1的平均胸径是超低亲子代LF2胸径的134.81%~138.32%;HF1平均树高是LF2的134.60%~150.00%。亲本龙15和1339取自双8区生长发育正常的植株。
-
文库构建、无参转录组测序以及随后的unigene功能注释、基因表达分析等项目的分析参见有关文献[12-17]。
实验流程按照Illumina公司提供的标准步骤执行。文库质检合格后,用Illumina Hiseq4000测序仪进行测序,测序策略为双末端测序,即高通量测序数据通常为2×150 bp。测序深度采用施季森等[8-10]杉木转录组测序的方法,为6G。原始RAW reads过滤后,使用Trinity软件对高质量数据[Clean reads]进行从头组装,即进行de novo拼接,获得unigenes和transcripts。随后进行功能注释:将unigenes序列分别比对到NCBI的数据库Nr、Pfam、KEGG(kyoto encyclopedia of genes and genomes)、Swiss-port、KOG及GO(gene ontology),获得与unigene相似性最高的序列,继而确定该unigene的功能和名称。
-
通过Illumina Hiseq4000测序获得的转录组测序数据,需经过以下几个步骤的生物信息学处理与分析[12-17],方能获得有意义的结果:(1)原始数据处理;(2)序列组装;(3)unigene序列的功能注释;(4)差异表达的unigene的热聚类分析等。除了样本针叶的采集和研究方案的制定外,整个项目中的测序和初步分析委托杭州联川生物技术股份有限公司完成。
unigene的表达量FPKM(Fragments per kb per million fragments),其计算公式:FPKM=106C/[(NL)/103],式中:FPKM是某个基因(A)的表达量,C是唯一比对到基因A的片段数,N是唯一比对到所有unigene的总片段数,L为unigene A的碱基数。
1.1. 研究材料
1.2. 研究方法
1.2.1. cDNA文库准备及RNA-seq测序
1.2.2. 序列比对及差异表达基因分析
-
杉木亲本龙15(P1)和1339(P2)、超亲子代HF1和低亲子代LF2,每个品种有3个生物学重复,12株样树(4个样本组),测序测得的原始测序序列(Raw reads)介于4.3E+07~5.2E+07;12个样本的Clean reads分别介于4.3E+07~5.2E+07。Clean reads有效数据占原始Raw reads的比例在98%以上。
12个样本的Phred数值大于Q20和Q30的碱基占总体碱基的百分比,分别介于98.20%~98.84%和95.07%~96.4%。原始测序序列中,碱基G和C的数量总和占总碱基数的百分比介于44.20%~45.08%之间。
12株样树测得的基因表达量在不同区间的分布接近正态分布。综合以上几个测序质量评价指标,说明12个样株的测序质量较高,能满足后续研究和数据分析的要求。
-
从测序数据的拼接结果可见(表 2),平均GC%达40.78%;当读长数达50%时,该读长的长度为1 214 pb。综合其它各项结果,可以得出测序数据的组装拼接十分成功。
项目Item 总数All 平均GC
Mean GC/%最小基因长度
Min length最大基因长度
Max length总组装基座
Total assembled bases读长数达50%时
读长长度N50/pb基因Gene 80 171 40.78 201 14 718 49 803 726 1 214 转录本Transcript 131 241 40.89 201 14 718 113 134 804 1 660 Table 2. Statistics of splicing results
-
对Clean reads在6个数据库进行BLASTX分析。将获得的基因分别在Swiss-prot、Nr、Pfam、KEGG、KOG和GO中注释,结果杉木亲代和子代共注释有80 171个基因(表 3)。Nr数据库的注释结果为32.50%,这远远低于通常报道的结果(56%)。Nr数据库的注释结果与注释软件、阈值等有关,也与基因的组装结果有关。
项目Item Swiss-prot Nr Pfam KEGG KOG GO 基因数
Gene_number16 837 26 055 20 239 9 162 21 059 15 088 比例
Percentage/%21.00 32.50 25.24 11.43 26.27 18.82 Table 3. Statistics of BLAST annotation of different database
杉木为无参测序,测得的数据通常采用BLASTX序列比对,观察表 3可以发现,各数据库所注释的基因比率大于100%,这表明有的基因同时在不同的数据库得到重复注释。由于针叶树已进行全基因组测序的树种太少,因而与农作物相比,针叶树种中挖掘出的基因也少。为了获得良好的测序结果,今后要加快针叶树尤其是杉木的全基因组测序研究。
-
基于转录组测序结果,绘制亲本和子代基因传递与表达韦恩图(图 1)。可以发现,将杂种和双亲相比,基因差异表达呈现以下5种模式:(1)双亲中可表达但杂种中不表达(双亲共沉默型);(2)只在双亲之一中表达,不在杂种中表达(亲本特异表达型);(3)只在杂种中表达,不在双亲中表达(杂种特异表达型);(4)在杂种和一个亲本中表达(单亲表达一致型);(5)在双亲和杂种中都表达。前4种模式属基因表达质的差异,即存在与缺失变异(Presence/absence variation,简称PAV),而第5种模式则属于基因表达量的差异。以上结果与徐进等[18]对鹅掌楸(Liridendron Chinense (Hemsl) Sarg.)、张小蒙等[19]对水稻(Oryzal sativa L.)及王章奎等[20]对小麦(Triticum aestivum L.)的研究结果基本一致,与张君等[21]对大豆(Glycine max (L.) Merr.)的研究结果完全一致。这表明亲本间存在差异基因与杂种优势有关。

Figure 1. A Venn diagram of the parent and progenies
-
从杉木4个样本组组间两两比较的差异基因表达分析结果(图 2)可以看出,P2VSP1、HF1VSP1、HF1VSP2、LF2VSHF1 4组差异表达基因不多,上调表达差异基因少于下调基因数;LF2VSP1、LF2VSP2两组间的表达差异基因多,但上调基因占多数;每组内的上调/下调基因数略有差别,但两者都有基因表达。这一结果与丁昌俊等[4]在美洲黑杨中的研究结果基本一致。龙15×1339这一杂交组合内,HF1是超亲子代,LF2是低亲子代,HF1较LF2表达差异基因的数量少,上调/下调基因比率不均匀、不平衡,这导致了HF1生长快,LF2生长慢。

Figure 2. Differentially expressed genes in different groups
-
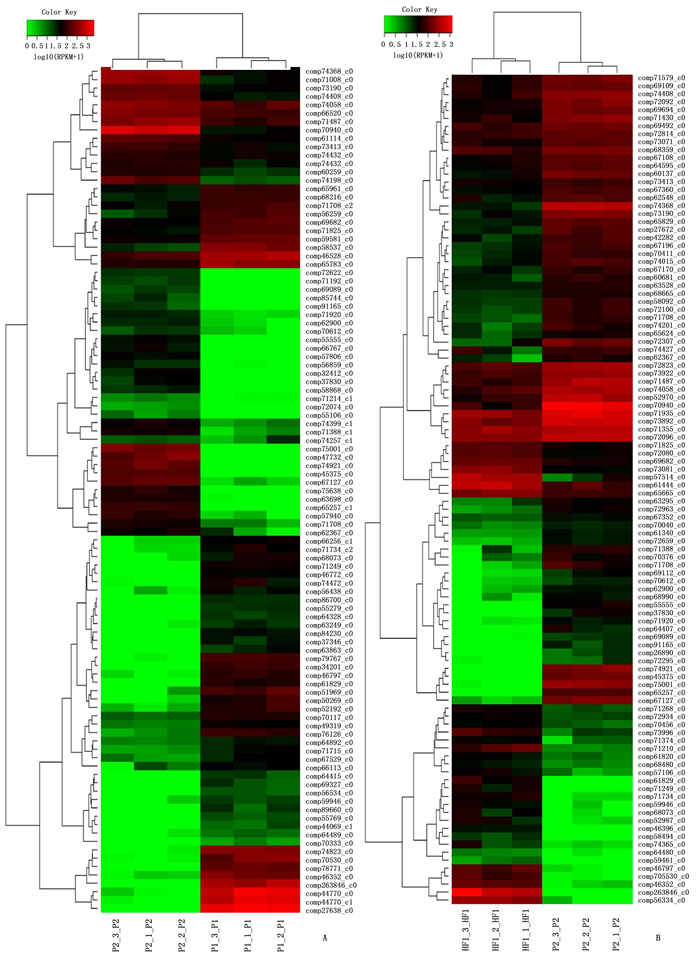
从测序组装、比对注释和基因表达量计算到基因的差异分析,最终从各样本组获得的基因中,依据显著富集的FDR值,分别抽取表达量极其显著的100多个基因。差异基因的富集分析是基于所有的差异基因进行的,并没有人为选择差异基因。根据基因表达量这一性状进行聚类分析,得到基因和亲本的聚类图(图 3)。从聚类图(图 3A)中可以发现:同一亲本不同分株在同一基因上通常表达量是一致的,100多个基因,按表达量聚类分为不同的聚类块。(1)由于同一聚类块的基因具有类似的功能,并假设它们控制一个性状,由于有些聚类块是由较少基因组成,有些聚类块的基因是多基因组成,这说明杉木数量性状有些是受寡基因控制,有些是受多基因控制的。(2)参与杂交的2个亲本,在相同的聚类块上,通常龙15的基因显著高表达,1339的同一基因则显著低表达,反之龙15的基因显著低表达,1339的同一基因则显著高表达,即同一基因在父母本间不同亲本基因表达量变化是互补的。既然龙15×1339这样组配产生了杂种优势,这表明杉木杂种(龙15×1339)生长性状的杂种优势是超显性在起作用(图 3 A)。(3)超亲子代和1339间,104基因按表达量聚类,获得10个聚类块(图 3B):Ⅰ类有16个基因,亲本和超亲子代同上调;Ⅱ类有18个基因,亲本上调,超亲子代下调;Ⅲ类有2个基因,亲子同为上调;Ⅳ类有10个基因,亲子同上调;Ⅴ类有7个基因,亲本下调,超亲子代上调;Ⅵ类有26个基因,亲本上调,子代下调;Ⅶ类有9个基因,亲本下调,子代上调;Ⅷ类有9个基因,亲本下调,子代上调;Ⅸ类有2个基因,亲子同为下调;Ⅹ类有5个基因,亲本下调,子代上调。这10个聚类块大致控制10个性状。其中,Ⅴ类、Ⅶ类、Ⅷ类和Ⅹ类这4个性状(聚类块)中,同一基因,亲本1339下调,超亲子代上调。
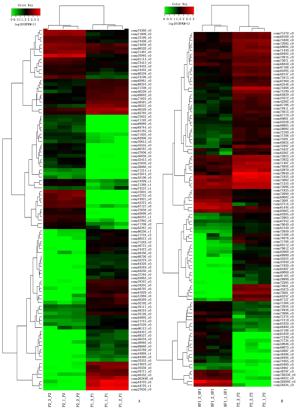
Figure 3. Hierarchical clustering analysis results were shown extremely significant difference between parent and offspring genes
对超亲子代与龙15(HF1VSP1)进行热聚类分析,聚类结果与HF1VSP2基本相同(图略),但使用的基因不同。100个基因,按表达量划分为7个聚类块,控制7个性状的表达:Ⅰ类有30个基因,亲本上调,子代下调;Ⅱ类有11个基因,亲本上调,子代下调;Ⅲ类有16个基因,亲子同为上调;Ⅳ类有5个基因,亲子同为下调;Ⅴ类有9个基因,亲本上调,子代下调;Ⅵ类有15个基因,亲子同为下调;Ⅶ类有14个基因,亲本下调,子代上调。可见,超亲子代与龙15的热聚分析中,只有Ⅶ类中的这14个基因可能与杉木杂交子代的高生产力有关,可能是控制生长性状杂种优势的因子。另外,作者还对亲本下调,超亲子代上调的基因进行了追踪,试图阐释子代表达量上调的这5个聚类块(性状)中的44个基因归属哪个代谢途径或参与了哪个性状的形成。超亲子代与龙15比较组Ⅶ类的14个基因追踪结果列于表 4。
基因Gene 基因的功能Gene function 基因Gene 基因的功能Gene function Comp73565_c0 与GO:0005634(细胞核)同源
Nuclear homologyComp72062_c3 [R]一般的功能预测
[R] Ggeneral functional predictionComp61820_c0 脂质运输和代谢
Lipid transport and metabolismComp71268_c0 昼夜节律Circadian rhythms Comp71030_c0 淀粉和蔗糖代谢
Starch and sucrose metabolismComp52987_c0 翻译后修饰;与叶绿体同源
Posttranslational modification; homologous to the chloroplastComp61016_c0 (只有上调表达量信息)
(Only upregulated expression information)Comp66767_c0 (只有上调表达量信息)
(Only upregulated expression information)Comp68480_c0 [R]一般的功能预测
[R] General functional predictionComp68783_c0 光合作用Photosynthesis Comp56601_c0 [P]无机离子运输和代谢
[P] Transport and metabolism of inorganic ionsComp70456_c1 昼夜节律;与GO:0030154同源
Circadian rhythms; same as GO: 0030154Comp62983_c0 苯丙氨酸代谢
Phenylalanine metabolismComp50961_c0 ATP结合;信号传递机制
ATP binding; signal transfer mechanismTable 4. 14 Genes function tracking results in HF1VSP1 Ⅶ class
可以看出,这14个在超亲子代中上调的基因,多数与生长有关:Comp71030_c0参与淀粉和蔗糖代谢;Comp62983_c0参与苯丙氨酸代谢;Comp71268_c0和Comp70456_c1参与昼夜节律;Comp68783_c0参与光合作用;Comp50961_c0参与ATP结合,并行使信号传递功能。显然这些基因与生长相关,是杉木高生产力的直接原因。
对于HF1VSP2,作者也对亲本下调,超亲子代中的上调基因进行了追踪:Ⅴ类的7个基因多与次生物代谢有关;Ⅶ类和Ⅷ类中的18个基因,有8个基因只有表达量信息,其余10个基因涉及不可或缺的膜、假设蛋白、翻译后修饰等功能;Ⅹ类的5个基因有3个只有表达量信息,另外2个基因涉及叶绿体被膜和一般功能预测;显然这4个聚类块(性状)与生长优势无直接关系,而是参与其它性状的形成。这些推断有待于今后杉木全基因组的测序和功能分析结果的验证。
2.1. 测序数据质量和基因表达概况
2.2. 测序数据的拼接结果
2.3. 基因注释结果
2.4. 亲代与其杂种子代4组样本间的基因差异模式分析
2.5. 亲代与其杂种子代4组样本间的差异基因表达分析

2.6. 基因表达量的聚类分析与杂种优势的分子机理
-
杂种优势已在许多作物中广泛利用,不少学者就杂种优势机理提出了许多假说,其中有代表性的假说有Bruce[22]提出的显性假说与Shull[23]提出的超显性假说。在杉木遗传育种领域,有关杉木生长性状也存在“加性学说”[24]和“显性学说”之争。随着分子生物学研究方法和技术的应用,植物遗传学研究不断拓展和深入,但有关杂种优势遗传机制的争议仍然存在,因为分子生物学研究结果仍与研究材料等因素有关[25-33]。如在水稻研究中,Xiao等[29]提出显性效应是水稻杂种优势的主要遗传基础,Yu等[30]则认为上位性效应是水稻杂种优势的重要遗传基础;庄杰云等[31]进一步考虑了遗传背景的影响,指出基于杂合型遗传背景的超显性效应是水稻杂种优势的重要遗传基础。
本研究得出杉木生长性状杂种优势的分子机理是超显性,这和本项目组关于杉木数量遗传的研究结果是一致的[2, 28]。随着杉木分子遗传学研究的深入,采用不同的杉木研究材料,使用不同的分析方法(如QTL分析),杉木数量性状的分子遗传机制的研究结果有可能与水稻中的情形相同或不同;也可能与本研究相同或不相同。杉木进入了高世代育种阶段,其亲本材料经过多世代与高强度选择,高世代育种群体在改良性状上多为杂合体,杂合体居多的育种群体,性状的遗传控制方式会由加性方差占主导地位,过渡到显性方差占主导地位[28]。同时杂种亲本间合理组配,会产生杂种优势。因此,杉木生长性状杂种优势分子机理为超显性这一结果,也适合于杉木高世代育种。
-
本研究采用转录组测序法研究杉木杂种优势的分子机理,得出杉木生长性状的杂种优势的分子机理是超显性。使用热聚类分析的方法,对基因和研究材料进行了类别划分,基因热聚类分析结果揭示,杉木有些性状通常由2~30个基因控制表达;杉木超亲子代高生产力的原因,是HF1VSP1中Ⅶ类中的14个基因在超亲子代中上调表达的结果。
韦恩图揭示杉木基因在上下代间的传递模式有5种:(1)双亲中可表达但杂种中不表达(双亲共沉默型);(2)只在双亲之一中表达,不在杂种中表达(亲本特异表达型);(3)只在杂种中表达,不在双亲中表达(杂种特异表达型);(4)在杂种和一个亲本中表达(单亲表达一致型);(5)在双亲和杂种中都表达。
通过杉木基因注释和功能分析,特别是热聚类分析,获得了诸如细胞膜、跨膜运输、细胞壁合成和防御反应的相关基因,以及植物昼夜节律、信号传导、淀粉和蔗糖代谢途径、苯丙素的生物合成代谢途径、激素合成与运输代谢等基因,这些上调/下调基因中有些基因参与了杂种优势的形成。
 DownLoad:
DownLoad: